Использование ML и новых методов кластеризации для борьбы с COVID-19: Революция в выявлении вирусных мутаций
Друзья, с момента основания проекта прошло уже 20 лет и мы рады сообщать вам, что сайт, наконец, переехали на новую платформу.
Какое-то время продолжим трудится на общее благо по адресу
На новой платформе мы уделили особое внимание удобству поиска материалов.
Особенно рекомендуем познакомиться с работой рубрикатора.
Спасибо, ждём вас на N-N-N.ru
Блог компании BotHub. Автор: Настасья Алексеевна. С момента появления COVID-19 мы застали множество “волн” и новых вспышек вируса. Помимо очевидной тяжести заболевания и невероятной скорости передачи, SARS-CoV-2 также отличается большим количеством различных мутаций, уклоняющихся от иммунных реакций.
Несмотря на то, что сейчас ситуация с коронавирусом стабилизировались, вирус продолжает развиваться и мутировать, что все равно сохраняет большую опасность для населения, в связи с трудностью и затратностью отслеживания его эволюции.
В данной статье я расскажу, как с помощью машинного обучения и новых методов кластеризации исследователям удалось встать на путь обнаружения новых вариантов вируса SARS-CoV-2, вызывающего COVID-19, со значительным временным и вычислительным выигрышем, по сравнению с существующими методами.
Введение в исследование
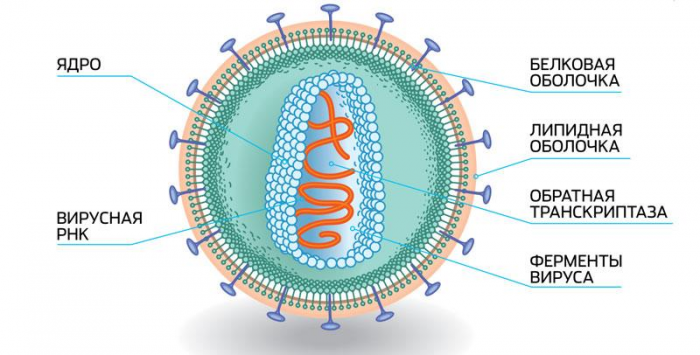
SARS-CoV-2 (вирус, вызывающий COVID-19), как и другие РНК-вирусы, имеет высокую скорость мутаций и короткое время генерации, что означает, что он эволюционирует так же быстро, как и распространяется. По этой причине филогенетический анализ стал мощным инструментом для отслеживания эволюции и распространения SARS-CoV-2.
РНК-вирусы — это вирусы, генетический материал которых представлен в виде РНК (рибонуклеиновая кислота), в отличие от более распространенных организмов, геном которых содержит ДНК (дезоксирибонуклеиновая кислота). РНК-вирусы включают в себя такие патогены, как вирус гриппа, вирус гепатита С, вирус ВИЧ, вирус гепатита B, коронавирусы (например, рассматриваемый SARS-CoV-2) и другие. Генетическая информация в РНК-вирусах может быть прямо использована для синтеза белков и для размножения вирусных частиц внутри клетки хозяина.
Филогенетический анализ — это изучение эволюции вида, группы организмов, геномов, белков и некодирующих последовательностей. С помощью филогенетического анализа ученые строят деревья родства, которые показывают, какие виды более тесно связаны и как происходило их эволюционное развитие. Этот метод позволяет понять и классифицировать организмы, а также изучать их эволюционные процессы.
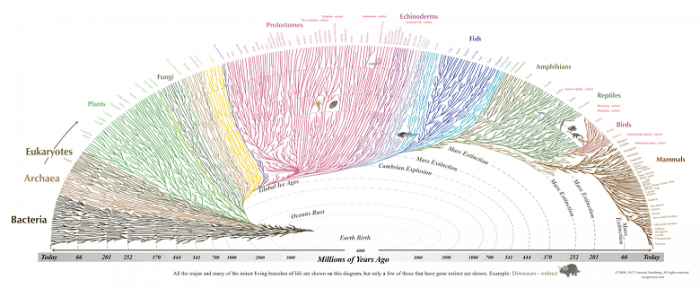 Филогенетическое дерево
Филогенетическое дерево
Большинство точечных мутаций, или однонуклеотидных полиморфизмов (SNP), которые появляются на филогенетических древах, нейтральны или почти нейтральны, что означает, что сами мутации практически не влияют на передачу и на то, будут ли линии расти или вымрут. Однако некоторые мутации действительно обладают избирательным преимуществом, поскольку они обеспечивают более эффективную передачу в популяции в момент появления.
Однонуклеотидные полиморфизмы (SNP) — это небольшие генетические изменения, которые происходят в одном нуклеотиде (базовой паре) ДНК. Например, если у одного человека в определенном месте генома может быть аденин (A), а у другого человека в том же месте — цитозин ©, то это будет представлять собой однонуклеотидный полиморфизм.
Одной из особенностей эволюции SARS-CoV-2 становится появление и распространение вариантов VOCs (Variants of Concern) и некоторых их основных сублиний, каждые из которых имеют большое количество провоцирующих линию мутаций (+которые обеспечили этим вирусным линиям преимущества в передаче).
VOCs – это варианты вируса, которые вызывают беспокойство из-за их способности передаваться легче, вызывать более тяжелые формы болезни или снижать эффективность вакцин.
Можно выделить наиболее значимые для распространения ковида:
- Alpha (B.1.1.7) – впервые обнаружен в Великобритании;
- Beta (B.1.351) – впервые обнаружен в Южной Африке;
- Gamma (P.1) – впервые обнаружен в Бразилии;
- Delta (B.1.617.2) – впервые обнаружен в Индии.
Выявление вирусных линий, которые могут стать проблематичными в будущем, требует значительных усилий, включая выравнивание последовательностей по некоторому эталону перед их включением в филогенетическое дерево; назначение новых линий (в частности, присвоение им номенклатуры, основанной на их местоположении); и идентификацию линий с потенциально проблемными мутациями.
Создание филогений, содержащих все высококачественные геномы SARS-CoV-2, могло бы помочь в автоматизации этого процесса, но при наличии почти 16 миллионов последовательностей, доступных в базе данных GISAID, выравнивание значительной части последовательностей и создание единой филогении возможно только при чрезвычайно больших вычислительных ресурсах.
В данном исследовании рассматриваются менее трудоемкие в вычислительном отношении методы неконтролируемого машинного обучения, которые могут работать с огромными объемами генетических данных и могут быть использованы для выявления растущих значимых линий.
- Источник(и):
- Войдите на сайт для отправки комментариев
